1. 基于生成式AI的蛋白从头设计
人工智能在酶工程领域的应用正在迅速发展,为研究人员提供了强大的工具,以更高效地探索蛋白质的功能与结构优化。通过深度学习和生成式AI模型,科学家可以从海量的天然蛋白质序列中识别进化规律,预测潜在的功能性变体,并在实验验证之前优化酶的活性和稳定性。尽管AI生成的蛋白序列尚未全面超越自然进化的成果,但其在加速筛选、提高工程效率以及降低实验成本方面展现出巨大潜力。未来,结合AI与实验技术的交叉研究,将有望推动酶工程迈向更高精度和可控性的新时代,为生物医药、绿色化学和可持续发展提供重要的技术支撑。
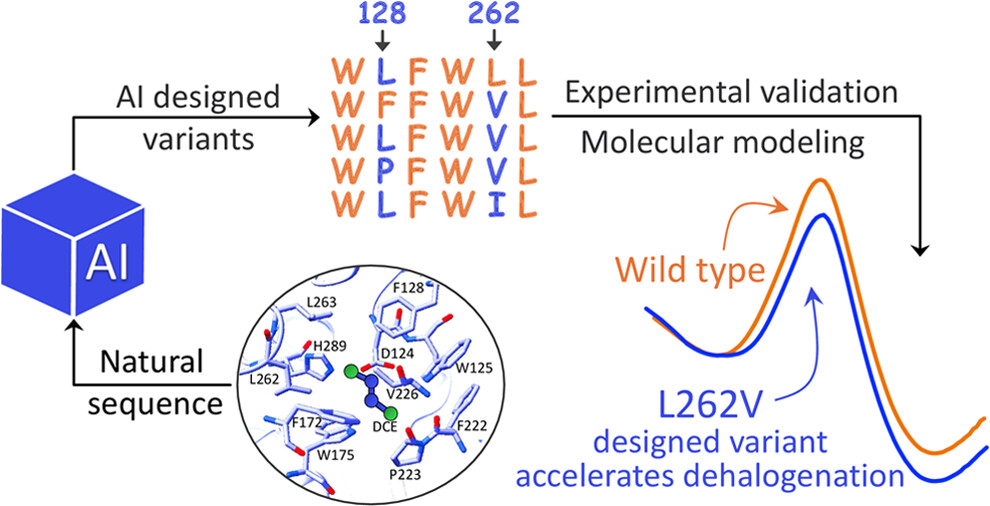
图 1. 蛋白设计的工作流程图。
相关论文:
1. Natalia Gelfand1, Vojtech Orel1, Wenqiang Cui1, Jiří Damborský, Chenglong Li, Zbyněk Prokop*, Wen Jun Xie*, and Arieh Warshel*, Biochemical and Computational Characterization of Haloalkane Dehalogenase Variants Designed by Generative AI: Accelerating the SN2 Step, Journal of the American Chemical Society, 2025, 147, 2747-2755.
2. 基于CADD和AIDD的药物设计
基于分子对接的虚拟筛选成本低、效率高,已成为药物发现的关键技术。分子对接采用的打分函数计算效率高但准确性相对较低,而且其往往忽略了靶蛋白柔性的影响。为提高虚拟筛选的预测精度,申请人围绕靶标-配体相互作用预测展开了深入系统的方法学研究,开发了一系列针对药物设计和筛选的计算工具和软件,并应用于多个重要靶标的药物研究。我们从2009年起对MM/PB(GB)SA方法中的若干核心理论问题进行了系统研究,深入探讨和评估了分子力场、溶剂化模型、原子电荷、构象熵、动力学采样时间等多种因素对MM/GBSA预测精度的影响,并针对不同体系提出了最优的预测策略 [1~11];针对传统MM/GBSA方法无法有效表征蛋白-配体相互作用界面的非均一极性环境的这一固有缺陷,创造性地发展了基于可变介电的VD-MM/GBSA结合自由能预测方法,其根据残基的物化性质和化学环境对其介电常数进行优化,有效提升了蛋白-配体结合自由能的预测精度 [12~13];在长期方法学研究的基础上,开发了CaFE、farPPI和HawkDock等一系列基于MM/PB(GB)SA方法的药物设计和筛选计算工具 [14~16]。
为了克服传统MM/GBSA方法体系依赖性的问题,发展了基于结合自由能分解和机器学习的MIEC-SVM方法,并成功应用于多肽-蛋白相互作用研究以及多个靶点的小分子抑制剂筛选 [17-18]。采用不同计算策略深入探讨了靶点构象变化对虚拟筛选的影响,证明了合理处理靶点柔性对准确预测抑制剂结合机制以及提升虚拟筛选精度的重要性;在此基础上提出了基于靶点多构象的整合虚拟筛选方法,并将其成功应用于ALK、AR、ROCK1等多个靶标的药物设计和筛选,证明通过整合靶点不同构象的预测结果能有效考虑靶点的柔性,从而显著提高虚拟筛选的预测精度(图3)[19-20]。
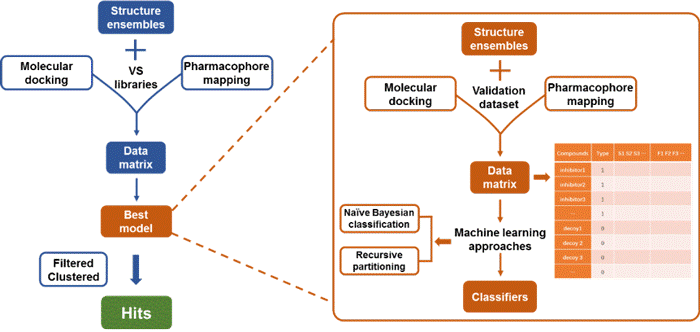
图 2. 整合的虚拟筛选策略流程图。
相关文献:
1. Wenqiang Cui and Shuguang Yuan*, Will the hype of automated drug discovery finally be realized?, Expert Opinion on Drug Discovery, 2024, 19, 259-262.
2. Wenqiang Cui, Qianwei Qu, Jinpeng Wang, Jinwen Bai, God’spower Bello-Onaghise, Yuang Li, Yonghui Zhou, Xingru Chen, Xin Liu, Sidi Zheng, Xiaoxu Xing, Nsabimana Eliphaz, and Yanhua Li*, Discovery of Potential Anti-infective Therapy Targeting Glutamine Synthetase in Staphylococcus xylosus, Frotiers in Chemistry, 2029, 7, 381.
3. 基于分子模拟的G蛋白偶联受体和酶蛋白的分子机理研究
先导化合物的发现和优化是新药开发中不可或缺的关键环节。计算机虚拟筛选(VS)技术能帮助我们有效发现结构新颖的活性分子,在药物开发的早期阶段发挥显著作用。我们课题组通过整合计算模拟以及体内/体外生物活性评价,针对多个重要药物靶点进行了先导化合物的筛选与优化。我们的优势在于广泛并深入涉足药物临床前开发的各个阶段,包括先导化合物的发现、化学合成优化、化学/生物活性检测、成药性和安全性评价等,能加快药物发现和开发的速度,并降低药物开发的成本。目前我们研究的药物靶点包括AR、GR、ALK、ROCK、PI3K、MIF、ALK、Tie-2和A2A等 [1~15]。例如:我们采用MIEC-SVM预测模型、分子对接、结合自由能计算等多种CADD方法设计并合成了一类新型的抗耐药性ALK “桥联”小分子,该类抑制剂可同时靶向ATP活性位点和邻近的疏水别构口袋;该类抑制剂在分子层面以及细胞层面均表现出优异的生物活性,是目前已报导的体外活性最强的I1/2型ALK抑制剂(001-017:IC50=0.27 nM),较已上市药物克唑替尼和色瑞替尼体外活性更好,其中001-017对多种ALK耐药突变体仍表现出很强的抑制活性及高度的激酶选择性,有望进入临床前研究 [12]。
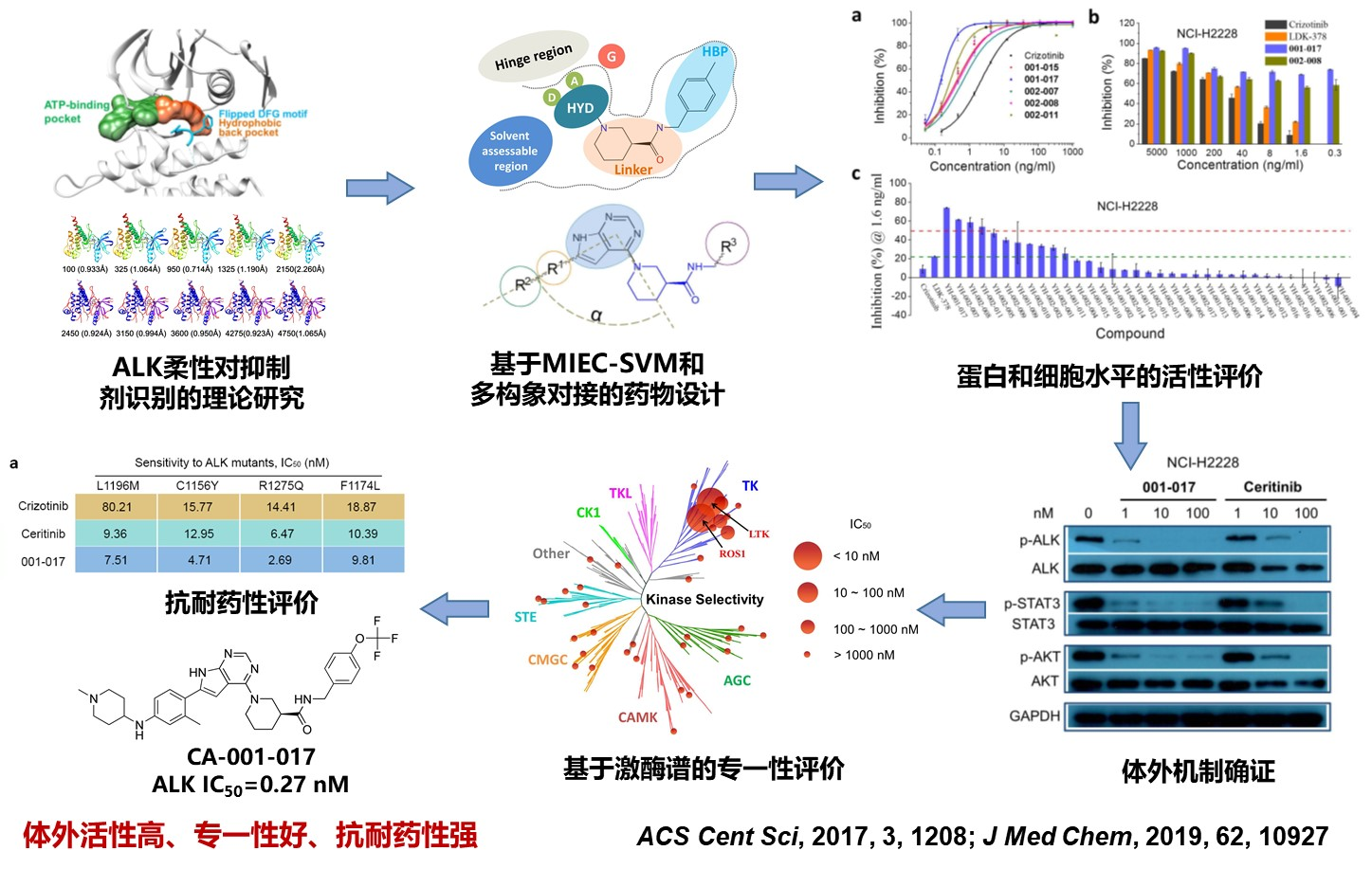
图 3. 新型ALK抑制剂的发现流程。
相关文献:
1. Wenqiang Cui, Junlin Dong, Shiyu Wang, Horst Vogel, Rongfeng Zou*, and Shuguang Yuan*, Molecular basis of ligand selectivity for melatonin receptors, RSC Advances, 2023, 13, 4422-4430.
2. Haijian Li1, Xiaolin Sun1, Wenqiang Cui1, Marc Xu1, Junlin Dong1, Babatunde Edukpe Ekundayo1, Dongchun Ni1, Zhili Rao1, Liwei Guo1, Henning Stahlberg*, Shuguang Yuan*, and Horst Vogel*, Computational drug development for membrane protein targets, Nature Biotechnology, 2024, 42, 229-242.